بدأت دراسة أمراض الميتوكوندريا الناجمة عن ضعف أكسدة بيتا للأحماض الدهنية ذات أطوال سلاسل الكربون المختلفة في عام 1976، عندما وصف العلماء لأول مرة المرضى الذين يعانون من نقص إنزيم أسيل-CoA ديهيدروجينيز للأحماض الدهنية متوسطة السلسلة وحمض الجلوتاريك من النوع الثاني.
يرتبط الممثلون الرئيسيون لهذه المجموعة من الأمراض بشكل أساسي بنقص إنزيمات الميتوكوندريا التالية: فوماراز، ومجمع ألفا كيتو غلوتارات ديهيدروجينيز، وسكسينات ديهيدروجينيز، وأكونيتاز.
من بين الأمراض الوراثية المُحددة لاستقلاب حمض البيروفيك، تُميّز عيوب مُركّب بيروفات ديهيدروجينيز وبيروفات كاربوكسيلاز. معظم هذه الحالات، باستثناء نقص مُكوّن ألفا E،
وُصف داء مينكيز (داء الشعر المجعد، OMIM 309400) لأول مرة من قِبل ج. هـ. مينكيز عام ١٩٦٢. يتراوح معدل الإصابة بالمرض بين ١:١١٤,٠٠٠ و١:٢٥٠,٠٠٠ مولود جديد. وهو وراثي متنحي مرتبط بالكروموسوم X.
وُصفت متلازمة ولفرام (متلازمة DIDMOAD - داء السكري الكاذب، داء السكري الميتيتوسي، ضمور العصب البصري، الصمم، OMIM 598500) لأول مرة من قِبل دي جي ولفرام وإتش بي واجينر بي عام ١٩٣٨ على أنها مزيج من داء السكري لدى الأطفال وضمور العصب البصري، والذي استكمل لاحقًا بداء السكري الكاذب وفقدان السمع. وحتى الآن، سُجلت حوالي ٢٠٠ حالة من هذا المرض.
وُصف اعتلال عضلي متعدد التصلب التقدمي لدى ألبرز (OMIM 203700) لأول مرة من قِبل بي. جيه. ألبرز عام ١٩٣١. لم يُحدَّد تواتر الإصابة به بعد. وهو وراثي جسمي متنحي. ولم يُحدَّد موقع الجين.
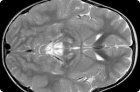
ذُكر هذا المرض لأول مرة عام ١٩٥١. وحتى الآن، سُجِّلت أكثر من ١٢٠ حالة. داء لي (OMIM ٢٥٦٠٠٠) هو مرض وراثي غير متجانس، يمكن أن يُورَّث إما نوويًا (جسميًا متنحيًا أو مرتبطًا بالكروموسوم X) أو ميتوكوندريا (أقل شيوعًا).